All published articles of this journal are available on ScienceDirect.

Molecular Epidemiology of Streptococcus equi subsp.zooepidemicus Isolated from Thoroughbred Horses Using Multi Locus Sequence Typing (MLST) in Korea
Abstract
Background:
Multi Locus Sequence Typing (MLST) is a new global molecular typing method used for analyzing the DNA polymorphisms in bacteria. In this study, using MLST, we analyzed the sequence profiles of Streptococcus (S.) zooepidemicus isolates from the Jeju and Jangsu provinces in South Korea.
Objective:
This study characterized the molecular epidemiology of S. zooepidemicus infection in Thoroughbred horses using MLST with an aim to control and prevent bacterial endometritis in mares.
Methods:
A total of 79 S. zooepidemicus isolates were included in this study. Sequencing of the 7 loci for the MLST analysis was performed as described in the MLST website manual (http:// pubmlst.org/szooepidemicus/) with some modifications. For each locus, every unique sequence was assigned a distinct allele number, and each Sequence Type (ST) was defined by a series of 7 integers (the allelic profile) corresponding to the alleles at the 7 loci (arcC, nrdE, proS, spi, tdk, tpi, and yqiL) using the MLST module in the Main Workbench.
Results:
Among the 79 isolates, 73 different MLST patterns were identified, each corresponding to 1-3 strains. The genetic relationships between the 79 isolates are presented in a dendrogram, and they diverged by up to 11% homology. At 11% homology, three MLST groups, M1, M2, and M3, were identified, and at 26% homology, five subgroups, L1-L5, were observed.
We observed various MLST patterns in the strains isolated from Jeju and Jangsu. In addition, by estimating the epidemiological relationships among the strains isolated from Jangsu in 2007 and Jeju in 2009, which had similar MLST patterns, we determined that some strains from Jangsu may have been transported to Jeju.
Conclusion:
MLST can be used as a framework for determining the epidemiological relationships that form the genetic basis of host and infection site selection.
1. INTRODUCTION
Bacterial infections are a frequent cause of infertility in mares, which is responsible for significant losses in the equine industry. A relatively wide range of microorganisms has been identified in endometrial swabs from mares with endometritis, and Streptococcus equi subsp. zooepidemicus (S. zooepidemicus) is the most frequently isolated pathogen [1-3].
S. zooepidemicus belongs to the β-hemolytic group C streptococci, which can cause disease in animals and humans [4]. S. zooepidemicus can be found in the nasopharynx, tonsils, respiratory tract, and on the genital mucous membrane of healthy horses and cattle. It is an important cause of respiratory tract infections in foals and young horses, and it is associated with uterine infections in mares [2]. It has also been associated with a wide variety of infections, including mastitis, in pigs, sheep, cows, goats, and several other mammalian species. Human infections are rare; however, the clinical signs in humans include pharyngitis, septicemia, meningitis, purulent arthritis, and endocarditis [2, 4-7].
Bain [8] reported that β-hemolytic Streptococcus was the most severe pathogen in the first year of infection, and only 40% of infected horses could deliver healthy foals and 35% of all infected horses could not achieve a successful pregnancy for 3 years. S. zooepidemicus causes 75% of vaginal infection and most septic abortions [9]. Furthermore, Escherichia coli, coagulase-positive Staphylococcus, Klebsiella pneumoniae, Pseudomonas aeruginosa, Corynebacterium equi, and fungal infections have also been shown to be severe pathogens resulting in difficult recoveries [10, 11]. Eliminating such pathogens from the genital tract is difficult, and the infections have been proven to be difficult to treat in some cases. However, the normal flora inhabiting the prepuce of horses and the external genitalia and vaginal vestibulum of mares can typically inhibit the growth of these virulent bacteria [8, 12].
The unambiguous characterization of the strains is crucial for addressing questions related to its epidemiology, population, and evolutionary biology. Multi Locus Sequence Typing (MLST), which characterizes strains using the sequences of 7 housekeeping loci, has become the method of choice for molecular typing of many bacterial and fungal pathogens as well as non-pathogens, and MLST schemes and strain databases are available for a growing number of prokaryotic and eukaryotic organisms [13]. Sequence data are ideal for strain characterization as they are unambiguous, meaning that strains can readily be compared between laboratories via the internet [13]. Laboratories undertaking MLST can quickly progress from sequencing the 7 gene fragments to characterizing the strains and determining their relationship to strains submitted by others and to the population as a whole. The MLST website provides a gateway to a number of MLST schemes, each of which contains a set of tools for the initial characterization and methods for relating query strains to other strains of the species, including clustering based on differences in allelic profiles, phylogenetic trees based on concatenated sequences, and a recently developed method, called eBURST, for identifying clonal complexes within a species and displaying the overall structure of the population. The address of the MLST website is http://www.mlst.net [14].
This study was prompted by our desire to determine the pathway of the disease that led to an abortion in an experimental mare at 5 months of pregnancy. Both the stallion and impregnated mare were imported into the Jeju province at the same time (in 2006) after following the quarantine procedures required to introduce a stallion from other countries. The disease inspection showed no unusual cause for the abortion in the mare or fetus. Finally, we confirmed the cause of the abortion to be an S. zooepidemicus infection.
The subtyping of S. zooepidemicus isolates is epidemiologically important for monitoring S. zooepidemicus infectious outbreaks in Thoroughbred horses. MLST is a new global molecular typing method used for analyzing the DNA polymorphisms in bacteria. In this study, using MLST, we analyzed the sequence profiles of S. zooepidemicus isolates from the Jeju and Jangsu provinces.
2. MATERIALS AND METHODS
2.1. Bacterial Isolates
A total of 79 S. zooepidemicus isolates were included in this study. All the isolates were recovered from the vaginal and uterine swabs of 78 mares with clinical signs of endometritis and 1 aborted fetus. The bacteria were isolated from clinical specimens on blood agar supplemented with nalidixic acid (15 mg/L). Molecular identification of the S. zooepidemicus isolates was performed by sodA–seeI specific multiplex PCR using the following primer pairs: SodA-F (5ʹ-CAG CAT TCC TGC TGA CAT TCG TCA GG-3ʹ) and SodA-R (5ʹ-CTG ACC AGC CTT ATT CAC AAC CAG CC-3ʹ); SeeI-F (5ʹ-GAA GGT CCG CCA TTT TCA GGT ACT TTG-3ʹ) and SeeI-R (5ʹ-GCA TAC TCT CTC TGT CAC CAT GTC CTG-3ʹ) [15]. Each PCR mixture contained 2 µL of DNA, 2 µL (10 pmol/µL) of each primer, 12.5 µL of 10× reaction buffer (Qiagen, Germany), and 6.5 µL of distilled water in a total volume of 25 µL. The PCR amplification program for sodA-seeI was as follows: an initial denaturation step at 94°C for 3 min, followed by 30 cycles of 94°C for 30 sec, 59°C for 30 sec, and 72°C for 40 sec. After identification, the bacteria were stored at -70°C until use [15].
2.2. Preparation of Chromosomal DNA
Overnight cultures of the bacteria were suspended in 300 µL of sterile, ultra-high quality water, heated on a heat block (Biometra, Germany) at 100°C for 10 min, and centrifuged at 10,000 × g (Thermo, USA) for 1 min. An aliquot of the supernatant (200 µL) containing genomic DNA was removed and placed into a fresh tube. The DNA was extracted from this sample using the Mag DNA extraction kit (Toyobo, Japan) according to the manufacturer’s instructions and then resuspended in 100 µL of sterile, ultra-high quality water. The DNA yield was approximately 2 µg/mL [16].
2.3. MLST Analysis
Sequencing of the 7 loci for the MLST analysis was performed as described in the MLST website manual (http:// pubmlst.org/szooepidemicus/) with some modifications. Internal fragments of carbamate kinase (arcC), ribonucleoside-diphosphate reductase (nrdE), prolyl-tRNA synthetase (proS), signal peptidase I (spi), thymidylate kinase (tdk), triosephosphate isomerase (tpi), and acetyl-CoA acetyltransferase (yqiL) were amplified by PCR from chromosomal DNA. The sequences of the oligonucleotide primers and the conditions for the PCR amplification and sequencing are presented in Tables 1 and 2.
| Target Gene | Primers Name | Sequence (5'-3') | PCR Program |
|---|---|---|---|
| arcC |
arcC1 arcC4 |
AGC CAT CTA CCG ACT AAC AC TCT GAA AGG GTT TGG CTA GC |
34×(94°C 30 s, 52°C 30 s, 72°C 1 min) |
| nrdE |
nrdE1 nrdE4 |
TTC TCC TTC AGG TGA CAG ATG AGA CTA GGC GTT TGA ACC TG |
34×(94°C 30 s, 54°C 30 s, 72°C 1 min) |
| proS |
proS1 proS4 |
TTG GTT GGA AAT GAC CAG ATC CCT GAT CCT TGA CAT TAA CGG |
34×(94°C 30 s, 51°C 30 s, 72°C 1 min) |
| spi |
spi1 spi4 |
CTT AGA GCA TTG CGC TAA GC TTG CCT GCT ATC TAG GGA AG |
34×(94°C 30 s, 50°C 30 s, 72°C 1 min) |
| tdk |
tdk1 tdk4 |
GAA TTC AGG AAA GAC CAT TG TAA TGC TTG CGA CAA ACT GG |
34×(94°C 30 s, 54°C 30 s, 72°C 1 min) |
| tpi |
tpi1 tpi4 |
GGC AGT AGT AAG CAA ATT ACC AGC AAG GCA AGG AAG CTA TC |
34×(94°C 30 s, 51°C 30 s, 72°C 1 min) |
| yqiL |
yqiL1 yqiL4 |
CCA CAT GGG AAT TAC AGC AG TCA ATA CAG ACG TCC CTT GAC C |
34×(94°C 30 s, 53°C 30 s, 72°C 1 min) |
| Target Gene | Primers Name | Sequence (5'-3') | PCR Program |
|---|---|---|---|
| arcC |
arcC2 arcC3 |
TGT CGC ACT TGG AGG AAA TG CAC CAC AAC ACC AGA ATC AAC |
1× (96 1min), 25×(96°C 10s, 50°C 5s, 72°C 4min) |
| nrdE |
nrdE2 nrdE3 |
CGC TCT ATT AAT TCT GCC TTG C GCA TAG GTT GCT CAT GAT GAT G |
|
| proS |
proS2 proS3 |
GAC TTC TTA GAG CCA GCT AG AAC GGA GCT AGC TCT TTA GG |
|
| spi |
spi2 spi3 |
CCT ACT TTT TGG ACT CTC ACG GAT TTT ATT GAG TGG CCA AAA GCG |
|
| tdk |
tdk2 tdk3 |
CTT GAA GGT AGC ACA CAA TTA TG GGT CTC ATT GCC ACC AAT TTG |
|
| tpi |
tpi2 tpi3 |
GGT CAC TAC AAT TGA GGC TG TTC AGG CTT CAC AGA ACC AC |
|
| yqiL |
yqiL2 yqiL3 |
AGT ACC GCA ACG TAA AGG TG GCC TGA CGC TTT TCC ATC TC |
The reactions were performed in a total volume of 20 µL using Pfu DNA polymerase (Qiagen, Germany), and the amplification conditions were 34 cycles of 94°C for 30 s, 50–55°C for 30 s, and 72°C for 1 min. The amplified DNA fragments were purified using the PCR purification kit (Qiagen), and the amplicons were sequenced in reactions containing 1 µL of PCR product (containing 3-10 ng/µL DNA), 1 µL of BigDye v3.1 (Applied Biosystems, USA), 2 µL of 5× buffer (Applied Biosystems), 5 µL of sterile water, and 1 µL of primer (3 pmol/µL). For each gene fragment, both strands were sequenced. The cycling protocol and sequencing reaction product clean-up are described in Table 2. The sequencing was performed using an ABI 3130xl DNA Analyzer (Applied Biosystems).
Sequence data were assembled using the Main Workbench (CLC bio, USA), and high quality double-stranded sequence data were used for further analysis. For each locus, every unique sequence was assigned a distinct allele number, and each Sequence Type (ST) was defined by a series of 7 integers (the allelic profile) corresponding to the alleles at the 7 loci, in the following order (alphabetical) arcC, nrdE, proS, spi, tdk, tpi, and yqiL using the MLST module in the Main Workbench. A dendrogram representing the genetic relationship between the 79 S. zooepidemicus strains was drawn using the InfoQuest Character Types Module (Bio-Rad) as previously described.
3. RESULTS
3.1. S. zooepidemicus Strains
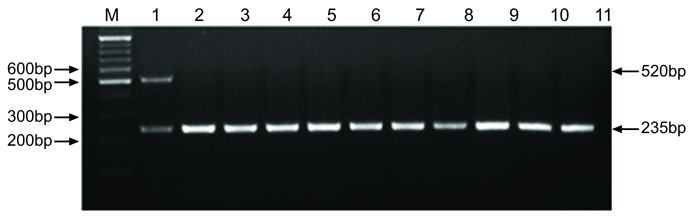
The species identification of the 79 strains was confirmed by a positive sodA (235 bp) reaction and a negative seeI (520 bp) reaction in the sodA-seeI multiplex PCR. The multiplex PCR used in this study has been previously shown to be a reliable molecular method to identify and differentiate S. zooepidemicus and S. equi (Fig. 1).
3.2. MLST
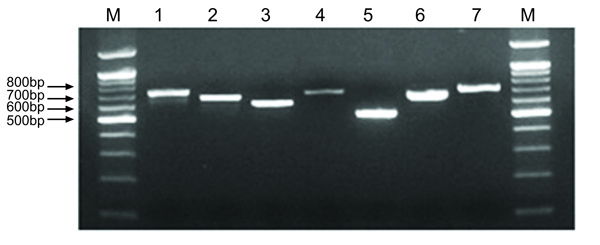
All 79 strains were positive for the 7 MLST alleles, with amplicon sizes of 500-800 bp. An agarose gel illustrating the typical amplicons for the 7 loci is shown in Fig. (2). The sequence data were assembled using Main Workbench, and high-quality double-stranded sequence data were used for further analysis (Fig. 3). All 73 of the STs found in the MLST analysis of the 79 isolates were new assignments. Of the new STs, 25 strains represented novel combinations of known alleles and new alleles (237, 243, 244, 246, 247, 250, 253, 254, 257, 258, 259, 264, 266, 268, 269, 270, 272, 284, 285, 295, 300, 301, 302, and 307). The new alleles included arcC 46-102, nrdE 48-107, proS 46-119, spi 46-120, tpi 46-110, tdk 46-114, and yqiL 44-121.
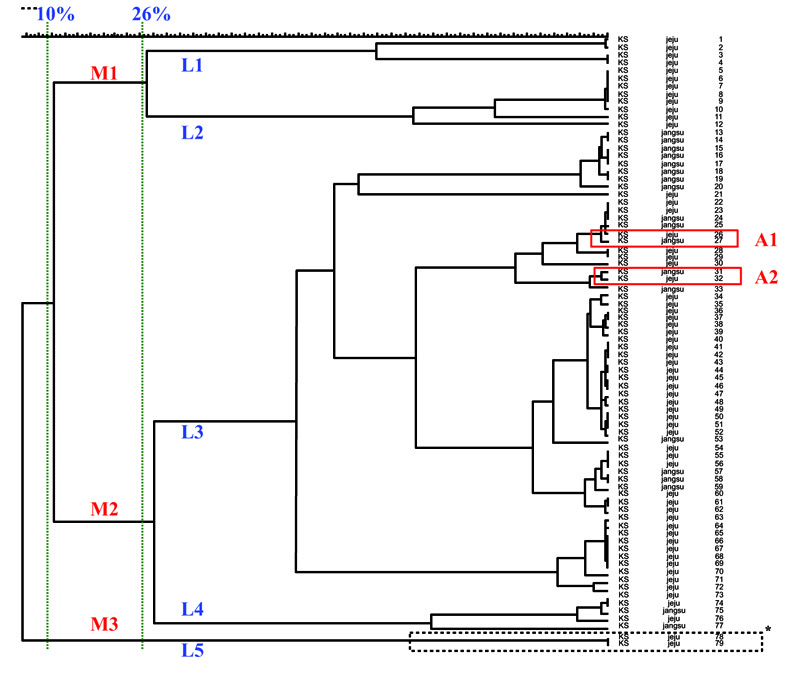
Among the 79 S. zooepidemicus isolates, 73 presented with different MLST patterns (Fig. 3), each corresponding to 1–3 strains. The genetic relationships between the 79 S. zooepidemicus isolates are presented in the dendrogram, and they diverged by up to 10% homology. At 10% homology, 3 MLST groups, M1, M2, and M3, were identified, and at 26% homology, 5 subgroups, L1-L5, were observed (Fig. 3).
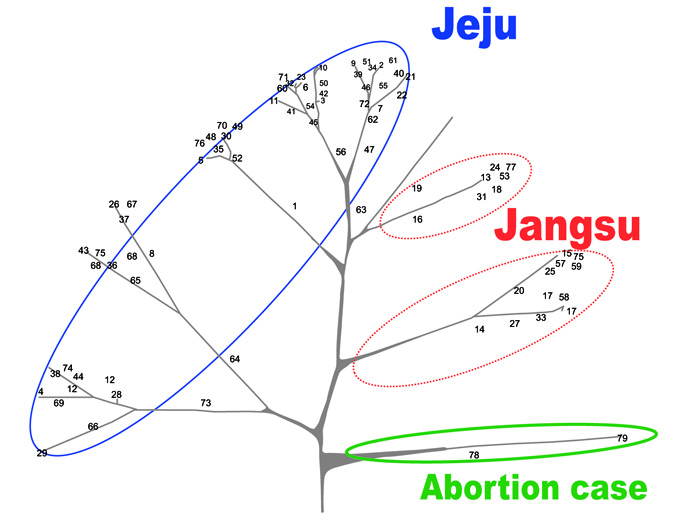
The results of the MLST analysis to compare the epidemiological properties of the S. zooepidemicus isolates from the abortion case (L5 subgroup) to other isolates from mares showed that the S. zooepidemicus isolates from both the mare that had the abortion and the aborted fetus exhibited the same pattern; however, these strains were not genetically similar to the strains isolated from other mares in Jeju or Jangsu (Fig. 4).
4. DISCUSSION
MLST is a well-established molecular typing method that has been used successfully to determine the population structure of many bacterial species [17-19]. Here, we report the development of an unambiguous and discriminatory MLST scheme for S. zooepidemicus. The data generated from the S. zooepidemicus isolates in this study have been used to construct a database on the main MLST website that permits direct comparison with data generated in other laboratories (http://pubmlst.org/szooepidemicus/).
PFGE is the gold standard for the subtyping of bacteria [20, 21]. However, in PFGE, various patterns can be observed in the analysis of the same strains depending on variations in the analysis, including the type of equipment, standard size marker, restriction enzyme, agarose concentration, and buffer concentration. Therefore, to overcome this weakness, world standardization is induced by organizing a global network; however, the established protocol is also limited to specific species, such as E. coli and Salmonella spp. Therefore, it is difficult to compare and analyze experimental data from unstandardized strains generated in different laboratories. For this reason, a new technique based on a standardized method that makes it possible to compare and analyze data between laboratories is greatly needed. Based on this need, the MLST technique was developed.
Data generated by MLST can not only be compared and analyzed to experimental data from the same laboratory but can also easily be compared to results from laboratories in other countries. Because the import and export of horses with predominant genetic traits are more common than that of other industrial animals, diseases occur more frequently in horses possibly because of disease spread as a result of extensive trade between countries. Therefore, analysis of the epidemiological characteristics of bacterial infectious diseases in horses must be accompanied by a comparison and analysis of the experimental data generated both within the same country and among countries.
Our analysis of the genetic features of S. zooepidemicus isolates from horse mares using MLST showed that in domestic isolates, allele types in the existing MLST database were identified; however, strains with identical genetic traits were not observed. In addition, in our analysis of 79 strains, 73 different MLST patterns were obtained, and efficient subgrouping was possible by analyzing the correlation among the 73 patterns. Based on these results, we judged that MLST of S. zooepidemicus strains is very efficient for identifying strains and is suitable for comparing and analyzing the epidemiological characteristics of strains isolated both within a single country and among different countries. However, the current S. zooepidemicus database has some weaknesses compared to those for Staphylococcus aureus, Yersinia spp., and Salmonella spp. strains; namely, the number of strains deposited in the database is lacking. If many more strains are added, it should be possible to compare and analyze worldwide experimental data.
The genetic relationship between S. zooepidemicus isolates based on the sequence patterns of 7 loci after the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) clustering is shown in Fig. (4). The vast majority of the MLST patterns (93.7%) were represented by a single isolate, and except for a cluster of 2 isolates with a similarity of 100%, the MLST patterns of the S. zooepidemicus isolates shared a low level of genetic similarity between them. These results showed the high genetic heterogeneity amongst the isolates studied, which may be related to either the wide distribution of this microorganism in the equine population or the fact that the S. zooepidemicus strains were isolated from individual mares, most of them belonging to single owners without any epidemiological or geographical relationship between them. These data demonstrate that many different strains of S. zooepidemicus, even from the same animal, could be isolated from uterine infections in mares. These findings correlate well with the fact that S. zooepidemicus is a mucosal commensal organism in the equine population, with the opportunistic character of this microorganism or possibility of the influx of disease from other areas, and the presence of isolated strains in a specific area for a long period of time.
Our genetic analysis using MLST to analyze the epidemiological characteristics of the S. zooepidemicus strains isolated from an abortion case showed that the S. zooepidemicus isolates from both the mare and aborted fetus had the same pattern; however, they were not genetically similar to other strains isolated from mares in Jeju or Jangsu (Fig. 4). Therefore, there is a good possibility that the strains isolated from the mare and aborted fetus were already present outside the country and that even if we exclude that possibility, the strains were not the same as other strains present in Jeju.
Different MLST patterns were observed by year and by region. However, the A1 and A2 subgroups, which showed similar patterns, included 2009 Jeju isolates and 2007 Jangsu isolates with 94% and 96% homology (Fig. 3). The phylogenetic analysis of the A1 and A2 subgroups showed that they have different genetic origins, with 85% homology, and the 4 isolates in the A1 and A2 groups have similar genetic origins. In addition, by estimating the epidemiological relationships among isolates from Jangsu in 2007 and Jeju in 2009, which had similar MLST patterns, we determined that some strains from Jangsu might have been imported into Jeju.
In our analysis of the genetic origins of the S. zooepidemicus strains isolated in Jeju from 2006 and 2009, we observed that the strains from 2006 and 2009 had different genetic origins. Based on the results, which showed that the strains from Jeju and Jangsu caused infections due to annual breeding, we believe that S. zooepidemicus strains come from many different areas and once established, can cause breeding-related diseases. As the breeding season ends, these strains disappear.
Studies on S. zooepidemicus isolates from horses in Asia are very limited compared with those in Europe and America. According to studies from these regions, S. zooepidemicus infection typically occurs on horse rearing farms and stud farms. Most infections occur through genital and respiratory infection and lead to fatal pneumonia, endometritis, and abortion.
CONCLUSION
There were no S. zooepidemicus studies specific to Asia. Therefore, to the best of our knowledge, this is the first study on S. zooepidemicus infection patterns and molecular epidemiological characteristics in Asia. Our report of the epidemiological characteristics of S. zooepidemicus isolates in Korea will be helpful for similar future studies. Furthermore, it will help not only the horses themselves but also the people who work on horse farms and at equestrian parks and animal hospitals by establishing an S. zooepidemicus screening system.
ETHICS APPROVAL AND CONSENT TO PARTICI PATE
The study received ethical approval from the College of Veterinary Medicine, Kyungpook National University.
HUMAN AND ANIMAL RIGHTS
The reported experiments were in accordance with the standards set forth in the 8th Edition of Guide for the Care and Use of Laboratory Animals (http://grants.nih.gov/grants/olaw /Guide-for-thecare-and-use-of-laboratory-animals.pdf) publi shed by the National Academy of Sciences, The National Academics Press, Washington DC, United States of America.
CONSENT FOR PUBLICATION
Not applicable
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
We thank the Ministry of Education, Science, Sports, and Culture (Monbukagakusho) Japan, for financial assistance.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

